Large-scale first principles configuration interaction calculations of optical absorption in aluminum clusters
Abstract
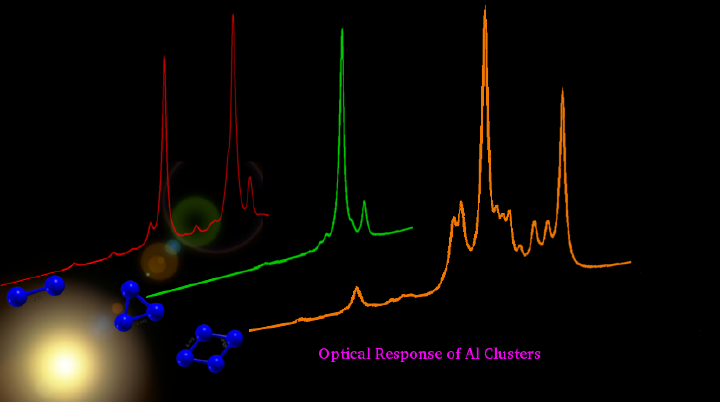
We report the linear optical absorption spectra of aluminum clusters Aln (n = 2-5) involving valence transitions, computed using the large-scale all-electron configuration interaction (CI) methodology. Several low-lying isomers of each cluster were considered, and their geometries were optimized at the coupled-cluster singles-doubles (CCSD) level of theory. With these optimized ground-state geometries, excited states of different clusters were computed using the multi-reference singles-doubles configuration-interaction (MRSDCI) approach, which includes electron correlation effects at a sophisticated level. These CI wave functions were used to compute the transition dipole matrix elements connecting the ground and various excited states of different clusters, and thus their photoabsorption spectra. The convergence of our results with respect to the basis sets, and the size of the CI expansion, was carefully examined. Our results were found to be significantly different as compared to those obtained using time-dependent density functional theory (TDDFT) [Deshpande et al. Phys. Rev. B Condens. Matter Mater. Phys., 2003, 68, 035428]. When compared to the available experimental data for the isomers of Al2 and Al3, our results are in very good agreement as far as important peak positions are concerned. The contribution of configurations to many body wave functions of various excited states suggests that in most cases optical excitations involved are collective, and plasmonic in nature.
Search content by categories
Ravindra Shinde
Research Scientist
I am a theoretical and computational condensed matter physicist and quantum chemist. I am currently working as a researcher at the University of Twente, the Netherlands. I am also the founder of The Science Dev.