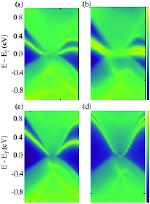
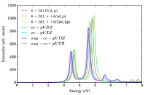
We have performed systematic large-scale all-electron correlated calculations on boron Bn, aluminum Aln and magnesium Mgn clusters (n=2–5), to study their linear optical absorption spectra. Several possible isomers of each cluster were considered, and their geometries were optimized at the coupled-cluster singles doubles (CCSD) level of theory. Using the optimized ground-state geometries, excited states of different clusters were computed using the multi-reference singles-doubles configuration interaction (MRSDCI) approach, which includes electron correlation effects at a sophisticated level. These CI wavefunctions were used to compute the transition dipole matrix elements connecting the ground and various excited states of different clusters, eventually leading to their linear absorption spectra. The convergence of our results with respect to the basis sets, and the size of the CI expansion was carefully examined. Isomers of a given cluster show a distinct signature spectrum, indicating a strong structure property relationship. This fact can be used in experiments to distinguish between different isomers of a cluster. Owing to the sophistication of our calculations, our results can be used for benchmarking of the absorption spectra and be used to design superior time-dependent density functional theoretical (TDDFT) approaches. The contribution of configurations to many-body wavefunction of various excited states suggests that in most cases optical excitations involved are collective, and plasmonic in nature. Optical absorption in planar boron clusters in wheel shape, B7, B8 and B9 computed using EOM-CCSD approach, have been compared to the results obtained from TDDFT approach with a number of functionals. This benchmarking reveals that range-separated functionals such as wB97xD and CAM-B3LYP give qualitatively as well as quantitatively the same results as that of EOM-CCSD.
 Illustration by Ravindra
Illustration by Ravindra